If you log in with SSH keys instead of a password, run kinit after connecting to access network storage (linuxhome, project storage). See Storage for details.
I know Linux but not clusters
→ Start with Slurm Basics
My code needs specific packages/versions
→ Read Apptainer to containerize your environment
I need to edit files on the cluster
→ Learn Vim for efficient editing over SSH
What you’ll be able to do
After completing these tutorials, you’ll be able to:
Log into DAIC and navigate the filesystem
Organize your projects with proper directory structures
Transfer data between your computer and the cluster
Submit batch jobs that run overnight
Request GPUs for deep learning training
Run parameter sweeps with job arrays
Package complex environments in containers
Edit files directly on the cluster
Getting help
Stuck on a command? Try man command or command --help
You’re a researcher who just got access to DAIC. You need to:
Set up a project directory
Organize your files
Find things when you forget where you put them
Automate repetitive tasks with scripts
Let’s learn the commands you need by actually doing these tasks.
Part 1: Finding your way around
When you log into DAIC, you arrive at your home directory. But where exactly are you, and what’s here?
Where am I?
The pwd command (print working directory) shows your current location:
$pwd/home/netid01
You’re in your home directory. On DAIC, this is a small space (5 MB) meant only for configuration files - not for your actual work.
What’s here?
The ls command lists what’s in the current directory:
$ ls
linuxhome
Not much! Let’s see more detail with ls -la:
$ ls -la
total 12
drwxr-xr-x 3 netid01 netid01 4096 Mar 20 09:00 .
drwxr-xr-x 100 root root 4096 Mar 20 08:00 ..
-rw-r--r-- 1 netid01 netid01 220 Mar 20 09:00 .bashrc
lrwxrwxrwx 1 netid01 netid01 45 Mar 20 09:00 linuxhome -> /tudelft.net/staff-homes-linux/n/netid01
Now we see hidden files (starting with .) and details about each file. The linuxhome entry has an arrow - it’s a symbolic link pointing to your larger personal storage.
Permission denied accessing linuxhome?
If you get “Permission denied” when accessing linuxhome, your Kerberos ticket has expired. Renew it with:
The cd command (change directory) moves you to a different location:
$cd linuxhome
$pwd/home/netid01/linuxhome
Some useful shortcuts:
$cd .. # Go up one level$cd ~ # Go to home directory$cd - # Go back to previous directory$cd# Also goes to home directory
Exercise 1: Explore the filesystem
Try these commands and observe what happens:
$cd /tudelft.net/staff-umbrella
$ ls
$cd ~
$pwd
Check your work
You should see project directories when listing /tudelft.net/staff-umbrella. After cd ~ and pwd, you should see your home directory path (e.g., /home/netid01).
Part 2: Understanding DAIC storage
Before we create files, let’s understand where to put them. DAIC has several storage locations:
Location
Purpose
Size
/home/<netid>
Config files only
5 MB
~/linuxhome
Personal files, code
~8 GB
/tudelft.net/staff-umbrella/<project>
Project data and datasets
Varies
Rule of thumb:
Code and small files → linuxhome or umbrella
Large datasets → umbrella
Never put large files in /home
Let’s navigate to where you’ll do most of your work:
$cd /tudelft.net/staff-umbrella
$ ls
You should see one or more project directories. For this tutorial, let’s assume you have access to a project called myproject:
The . means “start from current directory”. Common options:
$ find . -name "*.py"# Files matching pattern$ find . -type d -name "data*"# Directories only$ find . -type f -mtime -7 # Files modified in last 7 days$ find . -size +100M # Files larger than 100MB
Searching inside files
The grep command searches file contents:
$ grep "epochs" src/train.py
parser.add_argument('--epochs', type=int, default=10)
print(f"Training for {args.epochs} epochs with lr={args.lr}")
for epoch in range(args.epochs):
$ grep -n "epochs" src/train.py # Show line numbers$ grep -i "EPOCH" src/train.py # Case-insensitive$ grep -l "import" src/*.py # Just show filenames
Exercise 4: Find and search
Find all files modified in the last day:
$ find . -mtime -1
Search for all occurrences of “print” in your Python files:
$ grep -n "print" src/*.py
Find all directories named “data”:
$ find . -type d -name "data"
Check your work
The find . -mtime -1 command should list files you recently created. The grep -n command shows line numbers where “print” appears. The directory search should show ./data (and any other data directories you created).
Part 7: Automating with scripts
When you find yourself typing the same commands repeatedly, it’s time to write a script.
Your first script
Create a script that sets up a new experiment:
$ cat > setup_experiment.sh << 'EOF'#!/bin/bash
# Setup script for new experiments
# Check if experiment name was provided
if [ -z "$1" ]; then
echo "Usage: ./setup_experiment.sh <experiment_name>"
exit 1
fi
EXPERIMENT_NAME=$1
BASE_DIR="/tudelft.net/staff-umbrella/myproject"
echo "Creating experiment: $EXPERIMENT_NAME"
# Create directory structure
mkdir -p "$BASE_DIR/$EXPERIMENT_NAME"/{data,models,results,logs}
# Create a README
cat > "$BASE_DIR/$EXPERIMENT_NAME/README.md" << README
#$EXPERIMENT_NAMECreated: $(date)
Author: $(whoami)
## DescriptionTODO: Add description
## ResultsTODO: Add results
README
echo "Done! Experiment created at $BASE_DIR/$EXPERIMENT_NAME"
ls -la "$BASE_DIR/$EXPERIMENT_NAME"
EOF
Make it executable
Before you can run a script, you need to make it executable:
$ chmod +x setup_experiment.sh
$ ls -l setup_experiment.sh
-rwxr-xr-x 1 netid01 netid01 612 Mar 20 11:00 setup_experiment.sh
The x in the permissions means “executable”.
Run the script
$ ./setup_experiment.sh bert-finetuning
Creating experiment: bert-finetuning
Done! Experiment created at /tudelft.net/staff-umbrella/myproject/bert-finetuning
total 4
drwxr-xr-x 2 netid01 netid01 4096 Mar 20 11:00 data
drwxr-xr-x 2 netid01 netid01 4096 Mar 20 11:00 logs
drwxr-xr-x 2 netid01 netid01 4096 Mar 20 11:00 models
-rw-r--r-- 1 netid01 netid01 142 Mar 20 11:00 README.md
drwxr-xr-x 2 netid01 netid01 4096 Mar 20 11:00 results
Script building blocks
Here are patterns you’ll use often:
Variables:
NAME="experiment1"echo"Working on $NAME"
Conditionals:
if[ -f "data.csv"];thenecho"Data file exists"elseecho"Data file not found!"exit1fi
Loops:
for file in data/*.csv;doecho"Processing $file" python process.py "$file"done
Command substitution:
TODAY=$(date +%Y-%m-%d)echo"Running on $TODAY"
Exercise 5: Write a cleanup script
Create a script that removes old log files:
$ cat > cleanup_logs.sh << 'EOF'#!/bin/bash
# Remove log files older than 7 days
LOG_DIR="${1:-.}" # Use first argument, or current directory
echo "Cleaning logs in $LOG_DIR"
# Find and remove old logs
find "$LOG_DIR" -name "*.log" -mtime +7 -exec rm -v {} \;
echo "Done!"
EOF
$ chmod +x cleanup_logs.sh
$ ./cleanup_logs.sh logs/
Check your work
Verify the script is executable:
$ ls -l cleanup_logs.sh
-rwxr-xr-x 1 netid01 netid01 ... cleanup_logs.sh
The x in the permissions confirms it’s executable. When run, it prints “Cleaning logs in logs/” and “Done!” (plus any files it removes).
For more advanced shell customization, see Shell Setup.
2 - Slurm basics
Understanding the job scheduler on DAIC.
What you’ll learn
By the end of this tutorial, you’ll be able to:
Submit batch jobs that run on compute nodes
Request CPUs, memory, and GPUs for your jobs
Monitor job status and troubleshoot failures
Use interactive sessions for testing
Run parameter sweeps with job arrays
Time: About 45 minutes
Prerequisites: Complete the Bash Basics tutorial first, or be comfortable with Linux command line.
What is Slurm?
When you log into DAIC, you land on a login node. This is a shared computer where users prepare their work - but you shouldn’t run computations here. The actual computing happens on compute nodes, powerful machines with GPUs and lots of memory.
Slurm is the traffic controller that manages these compute nodes. When you want to run a computation, you don’t run it directly - you ask Slurm to run it for you. Slurm finds available resources, starts your job, and makes sure it doesn’t interfere with other users’ jobs.
Think of it like a restaurant: you don’t walk into the kitchen and cook your own food. You submit an order (your job), and the kitchen (Slurm) prepares it when they have capacity.
Why can’t I just run my code?
You might wonder: “Why can’t I just type python train.py and let it run?”
On a personal computer, that works fine. But DAIC is shared by hundreds of researchers, each wanting to use expensive GPUs. Without a scheduler:
Everyone would fight over the same resources
Your job might get killed when someone else starts theirs
GPUs would sit idle when no one happens to be logged in
There would be no fairness - whoever types fastest wins
Slurm solves these problems by:
Queueing jobs and running them in order
Guaranteeing that your job gets the resources you requested
Ensuring fair access based on policies
Maximizing utilization of expensive hardware
The two ways to run jobs
Batch jobs: submit and walk away
Most of the time, you’ll use batch jobs. You write a script that describes what you want to run, submit it, and Slurm runs it whenever resources are available. You don’t need to stay logged in - you can submit at 5pm, go home, and check results the next morning.
$ sbatch my_job.sh
Submitted batch job 12345
Your job enters a queue. When resources become available, Slurm runs it. Output goes to a file you can read later.
Interactive jobs: real-time access
Sometimes you need to work interactively - debugging, testing, or exploring data. For this, you request an interactive job. Slurm allocates resources, and you get a shell on a compute node.
Interactive jobs are great for testing but expensive - you’re reserving resources the whole time, even if you’re just thinking. Use batch jobs for actual computations.
Your first batch job
Let’s walk through creating and submitting a batch job step by step.
Step 1: Create a Python script
First, create a simple script to run. This one just prints some information:
$cd /tudelft.net/staff-umbrella/<project>
$ vim hello.py
importsocketimportosprint(f"Hello from {socket.gethostname()}")print(f"Job ID: {os.environ.get('SLURM_JOB_ID','not in slurm')}")print(f"CPUs allocated: {os.environ.get('SLURM_CPUS_PER_TASK','unknown')}")
Step 2: Create a batch script
Now create the Slurm script that will run your Python code:
$ vim hello_job.sh
#!/bin/bash
#SBATCH --account=<your-account>#SBATCH --partition=all#SBATCH --time=0:10:00#SBATCH --ntasks=1#SBATCH --cpus-per-task=1#SBATCH --mem=1G#SBATCH --output=hello_%j.outecho"Job started at $(date)"srun python hello.py
echo"Job finished at $(date)"
Let’s understand each line:
Line
Purpose
#!/bin/bash
This is a bash script
#SBATCH --account=...
Which account to bill (required)
#SBATCH --partition=all
Which group of nodes to use
#SBATCH --time=0:10:00
Maximum runtime: 10 minutes
#SBATCH --ntasks=1
Run one task
#SBATCH --cpus-per-task=1
Use one CPU core
#SBATCH --mem=1G
Request 1 GB of memory
#SBATCH --output=hello_%j.out
Where to write output (%j = job ID)
srun python hello.py
The actual command to run
Step 3: Find your account
Before submitting, you need to know your account name:
$ sacctmgr show associations user=$USERformat=Account -P
Account
ewi-insy-reit
Replace <your-account> in your script with this value (e.g., ewi-insy-reit).
Step 4: Submit the job
$ sbatch hello_job.sh
Submitted batch job 12345
The number 12345 is your job ID. You’ll use this to track your job.
Step 5: Check job status
$ squeue -u $USER JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
12345 all hello_jo netid01 PD 0:00 1 (Priority)
The ST column shows the status:
PD = Pending - waiting in queue
R = Running
CG = Completing - wrapping up
The REASON column tells you why a job is pending:
Priority = other jobs are ahead of you in the queue
Resources = waiting for nodes to become free
QOSMaxJobsPerUserLimit = you’ve hit your job limit
Step 6: Check the output
Once the job completes, read the output file:
$ cat hello_12345.out
Job started at Fri Mar 20 10:15:32 CET 2026
Hello from gpu23.ethernet.tudhpc
Job ID: 12345
CPUs allocated: 1
Job finished at Fri Mar 20 10:15:33 CET 2026
Your code ran on gpu23, not on the login node. Slurm handled everything.
Understanding resource requests
The most confusing part of Slurm is figuring out what resources to request. Request too little and your job crashes; request too much and you wait longer in the queue.
Time (--time)
How long your job will run. Format: D-HH:MM:SS or HH:MM:SS
#SBATCH --time=0:30:00 # 30 minutes#SBATCH --time=4:00:00 # 4 hours#SBATCH --time=1-00:00:00 # 1 day#SBATCH --time=7-00:00:00 # 7 days (maximum on DAIC)
Important: If your job exceeds this time, Slurm kills it. But requesting more time means waiting longer in the queue. Start with a generous estimate, then use seff on completed jobs to tune it.
NumPy/Pandas with parallelism: however many threads you configure
GPUs (--gres)
Request GPUs with the --gres (generic resources) option:
#SBATCH --gres=gpu:1 # One GPU (any type)#SBATCH --gres=gpu:2 # Two GPUs#SBATCH --gres=gpu:l40:1 # Specifically an L40 GPU#SBATCH --gres=gpu:a40:2 # Two A40 GPUs
Available GPU types on DAIC include L40, A40, and RTX Pro 6000. Request specific types only if your code requires it - being flexible gets you through the queue faster.
Running GPU jobs
Most deep learning jobs need GPUs. Here’s a complete example:
#!/bin/bash
#SBATCH --account=<your-account>#SBATCH --partition=all#SBATCH --time=1:00:00#SBATCH --ntasks=1#SBATCH --cpus-per-task=4#SBATCH --mem=16G#SBATCH --gres=gpu:1#SBATCH --output=train_%j.out# Clean environment and load required modulesmodule purge
module load 2025/gpu cuda/12.9
# Print job info for debuggingecho"Job ID: $SLURM_JOB_ID"echo"Running on: $(hostname)"echo"GPUs: $CUDA_VISIBLE_DEVICES"echo"Start time: $(date)"# Run trainingsrun python train.py
echo"End time: $(date)"
Understanding the module system
DAIC uses an environment modules system to manage software. Instead of having every version of every library available at once (which would cause conflicts), software is organized into modules that you load when needed.
The module commands set up your software environment:
module purge # Clear any previously loaded modulesmodule load 2025/gpu # Load the 2025 GPU software stackmodule load cuda/12.9 # Load CUDA 12.9
Why use modules?
Version control: Run module load python/3.11 today, python/3.12 tomorrow
Avoid conflicts: Different projects can use different library versions
Clean environment: module purge gives you a fresh start
If you forget, you’ll hold resources for the full time you requested, even if you’re not using them. This isn’t fair to other users.
Job arrays: running many similar jobs
Often you need to run the same code with different parameters - different random seeds, different hyperparameters, or different data splits. Job arrays make this easy.
The problem
You want to run your experiment with seeds 1 through 10. You could submit 10 separate jobs:
$ sbatch experiment_array.sh
Submitted batch job 12360
$ squeue -u $USER JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
12360_1 all experime netid01 R 0:30 1 gpu01
12360_2 all experime netid01 R 0:30 1 gpu02
12360_3 all experime netid01 R 0:30 1 gpu03
12360_4 all experime netid01 PD 0:00 1 (Resources)
...
Array variations
#SBATCH --array=1-100 # Tasks 1 through 100#SBATCH --array=0-9 # Tasks 0 through 9#SBATCH --array=1,3,5,7 # Just these specific tasks#SBATCH --array=1-100%10 # 1-100, but max 10 running at once
The %10 syntax limits concurrent tasks, useful if you don’t want to flood the queue.
Using array indices creatively
Your Python code can use $SLURM_ARRAY_TASK_ID for more than just seeds:
importosimportjsontask_id=int(os.environ.get('SLURM_ARRAY_TASK_ID',0))# Load hyperparameter configurationswithopen('configs.json')asf:configs=json.load(f)config=configs[task_id]print(f"Running with config: {config}")
Job 12392 waits for both 12390 and 12391 to complete.
Checking job history and efficiency
View past jobs
$ sacct -u $USER --starttime=2026-03-01
JobID JobName Partition Account AllocCPUS State ExitCode
------------ ---------- ---------- ---------- ---------- ---------- --------
12340 training all ewi-insy 8 COMPLETED 0:0
12341 failed all ewi-insy 4 FAILED 1:0
12342 training all ewi-insy 8 TIMEOUT 0:0
Exit codes:
0:0 = success
1:0 = your code exited with error
0:9 = killed by signal 9 (often out of memory)
TIMEOUT = exceeded time limit
Check efficiency
The seff command shows how well you used the resources you requested:
$ seff 12340Job ID: 12340
Cluster: daic
State: COMPLETED
Nodes: 1
Cores per node: 8
CPU Utilized: 06:30:15
CPU Efficiency: 81.3% of 08:00:00 core-walltime
Job Wall-clock time: 01:00:00
Memory Utilized: 24.5 GB
Memory Efficiency: 76.6% of 32.0 GB
This job used 81% of allocated CPU and 77% of allocated memory - reasonable efficiency. If you see numbers below 50%, you’re requesting more than you need.
Adjusting based on efficiency
If seff shows:
Low CPU efficiency: Reduce --cpus-per-task
Low memory efficiency: Reduce --mem
Very high efficiency (>95%): Consider requesting slightly more headroom
Troubleshooting
Job stuck in pending
Check why with squeue:
$ squeue -u $USER JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
12345 all training netid01 PD 0:00 1 (Resources)
Common reasons:
Priority - Other jobs are ahead of you. Wait, or request fewer resources.
Resources - Not enough free nodes. Wait, or request fewer resources.
QOSMaxJobsPerUserLimit - You’ve hit your concurrent job limit. Wait for some to finish.
AssocMaxJobsLimit - Your account has hit its limit.
Job killed immediately
Check the output file for errors. Common issues:
Out of memory:
slurmstepd: error: Detected 1 oom-kill event(s) in step 12345.0
Solution: Increase --mem
Time limit:
slurmstepd: error: *** JOB 12345 ON gpu01 CANCELLED AT 2026-03-20T12:00:00 DUE TO TIME LIMIT ***
Solution: Increase --time or add checkpointing to your code
Module not found:
ModuleNotFoundError: No module named 'torch'
Solution: Add module load commands to your script
Can’t find GPUs
Your code can’t see GPUs even though you requested them:
torch.cuda.is_available()# Returns False
Common causes:
Forgot --gres=gpu:1 in your script
Running on login node instead of through srun
Missing module load cuda
CUDA version mismatch
Best practices
1. Test before submitting long jobs
$ salloc --time=0:30:00 --gres=gpu:1 ...
$ srun python train.py --max-epochs 1# Quick test$exit$ sbatch full_training.sh # Now submit the real job
2. Request only what you need
Larger requests wait longer in the queue. Start small and increase if needed.
3. Use meaningful job names
#SBATCH --job-name=bert-finetune-lr001
Makes squeue output much more readable.
4. Save checkpoints
For long jobs, save state periodically so you can resume if killed:
# Save checkpoint every epochtorch.save({'epoch':epoch,'model_state_dict':model.state_dict(),'optimizer_state_dict':optimizer.state_dict(),},f'checkpoint_epoch_{epoch}.pt')
5. Use job arrays instead of many scripts
One job array is easier to manage than 100 separate submissions.
6. Check efficiency and tune
After your first successful run, check seff and adjust requests.
Quick reference
Submit and monitor
Command
Purpose
sbatch script.sh
Submit batch job
salloc ...
Request interactive session
srun command
Run command on allocated nodes
squeue -u $USER
View your jobs
scancel 12345
Cancel a job
scancel -u $USER
Cancel all your jobs
Information
Command
Purpose
sinfo
View partitions and nodes
scontrol show job 12345
Detailed job info
sacct -u $USER
View job history
seff 12345
Check job efficiency
sacctmgr show assoc user=$USER
View your accounts
Common sbatch options
Option
Example
Purpose
--account
ewi-insy
Billing account
--partition
all
Node group
--time
4:00:00
Time limit
--cpus-per-task
8
CPU cores
--mem
32G
Memory
--gres
gpu:1
GPUs
--output
log_%j.out
Output file
--array
1-10
Job array
Summary
You’ve learned:
Concept
Key Commands
Submit a batch job
sbatch script.sh
Request interactive session
salloc --time=1:00:00 --gres=gpu:1 ...
Run on allocated node
srun python train.py
Check job status
squeue -u $USER
Cancel a job
scancel <jobid>
View job history
sacct -u $USER
Check efficiency
seff <jobid>
Run parameter sweep
#SBATCH --array=1-10
Chain jobs
--dependency=afterok:<jobid>
Exercises
Try these on your own to solidify your understanding:
Exercise 1: Basic job submission
Create and submit a job that prints your username, hostname, and current date. Check the output.
Check your work
Your output file should contain something like:
netid01
gpu15.ethernet.tudhpc
Fri Mar 20 10:30:00 CET 2026
The hostname should be a compute node (not daic01).
Exercise 2: GPU job
Modify the basic job to request a GPU. Add nvidia-smi to verify the GPU is available.
Check your work
Your output should include nvidia-smi output showing a GPU:
+-----------------------------------------------------------------------------+
| NVIDIA-SMI ... Driver Version: ... CUDA Version: ... |
|-------------------------------+----------------------+----------------------+
| GPU Name ...
If you see “NVIDIA-SMI has failed”, check that you requested a GPU with --gres=gpu:1.
Exercise 3: Resource tuning
Submit a job, then use seff to check its efficiency. Was your resource request appropriate?
Check your work
Run seff <jobid> after your job completes. Good efficiency looks like:
CPU Efficiency: 70-95%
Memory Efficiency: 50-90%
If efficiency is below 50%, reduce your request next time.
Exercise 4: Job array
Create a job array that runs 5 tasks. Each task should print its array task ID.
Check your work
You should see 5 output files (e.g., job_12345_1.out through job_12345_5.out). Each should contain its task ID:
Containerization packages your software, libraries, and dependencies into a single portable unit: a container. This makes your application behave the same way everywhere: on your laptop, in the cloud, or on DAIC. This means:
Consistency: The application runs the same way regardless of where it’s executed. You can develop on one machine, test on another, and deploy on a cluster without worrying about dependency differences.
Isolation: Each container is independent from others, preventing conflicts and enhancing security and reliability.
Portability: Containers can run on different systems without modification, simplifying movement between servers, clusters, or clouds.
Efficiency: Containers share the host system’s resources like the operating system, making them lightweight and fast to start compared to virtual machines.
On DAIC specifically, users often encounter issues with limited home directory space or Windows-based /tudelft.net mounts (see Storage), which can complicate the use of conda/mamba and/or pip. Containers offer a solution by encapsulating all software and dependencies in a self-contained environment. You can, for instance, store containers on staff-umbrella with all required dependencies, including those installed via pip, and run them reliably and reproducibly without being limited by home directory size or mount compatibility.
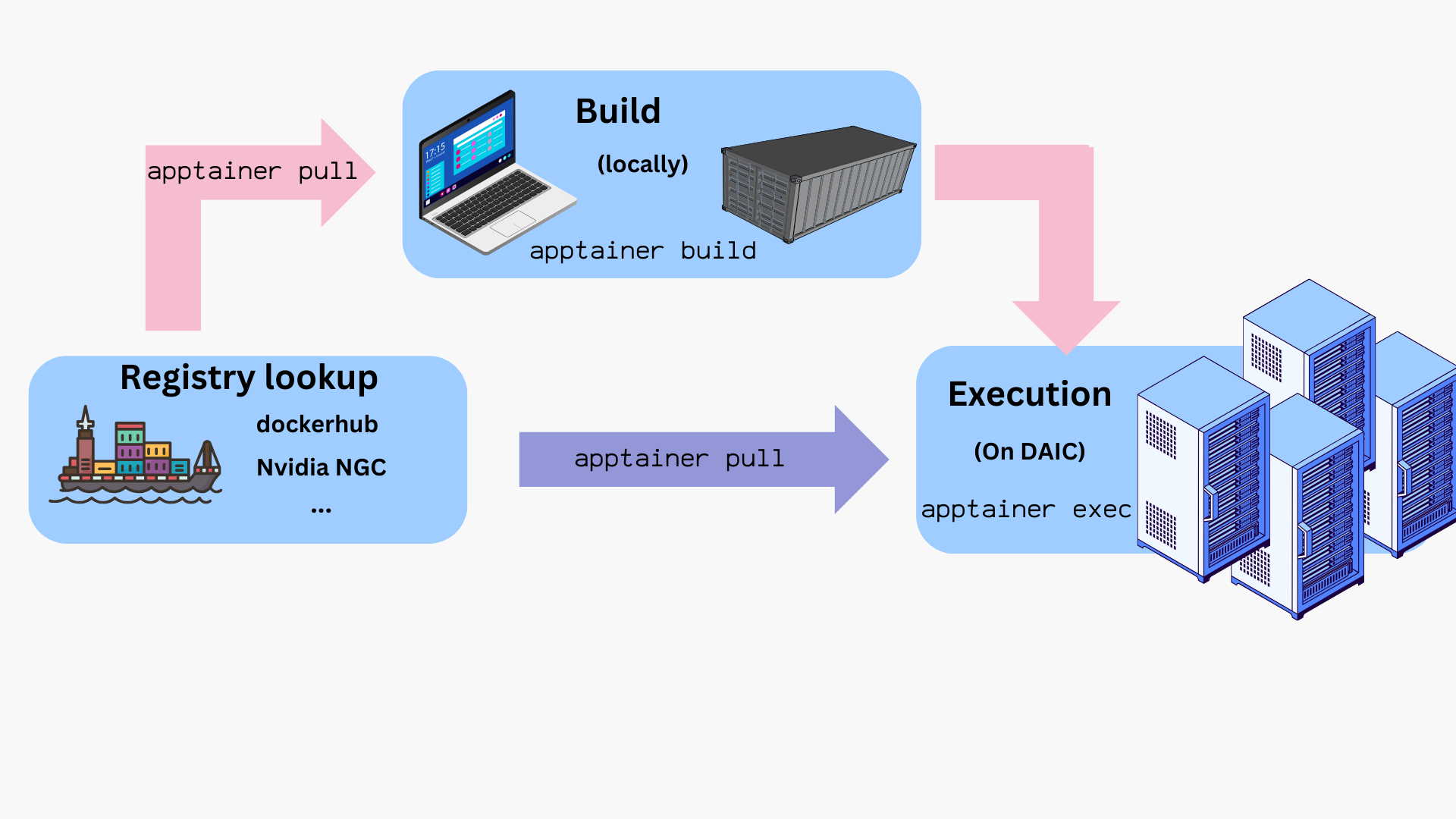
Containerization on DAIC: Apptainer
DAIC supports Apptainer (formerly known as Singularity), an open-source container platform designed for high-performance computing environments. Apptainer runs container images securely on shared clusters and allows you to use Docker images directly, without needing Docker itself.
A typical Apptainer workflow revolves around three key components:
Component
Description
Definition file (*.def)
A recipe describing how to build the container: which base image to use and which packages to install.
Image (*.sif)
A single portable file containing the full environment: operating system, libraries, and applications.
Container
A running instance of an image, with its own writable workspace for temporary files or intermediate data.
Because Apptainer integrates well with Slurm, containers can be launched directly within batch jobs or interactive sessions on DAIC. The following sections show how to obtain, build, and run images.
Workflow overview
The typical lifecycle for containers on DAIC is:
Build the image locally from a .def file.
Transfer or pull the resulting .sif file onto DAIC.
Test interactively using salloc to get a compute node.
Run in a batch job with sbatch or srun using apptainer exec or apptainer run.
Provision bind mounts, GPU flags, and cache locations as needed.
Clean up and manage storage (e.g., APPTAINER_CACHEDIR).
How to run commands/programs inside a container?
Once you have a container image (e.g., myimage.sif), you can launch it in different ways depending on how you want to interact with it:
Command
Description
Example
apptainer shell <image>
Start an interactive shell inside the container.
apptainer shell myimage.sif
apptainer exec <image> <command>
Run the <command> inside the container, then exit.
apptainer exec myimage.sif python --version
apptainer run <image>
Execute the container’s default entrypoint (defined in its recipe).
apptainer run myimage.sif
where:
<image> is the path to a container image, typically, a *.sif file.
Tips:
Use shell for exploration or debugging inside the container.
Use exec or run for automation, workflows, or Slurm batch jobs.
$ ls
ubuntu_latest.sif
$ apptainer exec ubuntu_latest.sif cat /etc/os-release
PRETTY_NAME="Ubuntu 22.04.2 LTS"
NAME="Ubuntu"
VERSION_ID="22.04"
...
$ ls /.apptainer.d/
ls: cannot access /.apptainer.d/: No such file or directory
$ apptainer shell ubuntu_latest.sif
Apptainer> hostname
daic01.hpc.tudelft.nl
Apptainer> ls /.apptainer.d/
Apptainer actions env labels.json libs runscript startscript
Apptainer> exit
Notes:
Inside the container, the command prompt changes to Apptainer>
The container inherits your environment (e.g., $HOME, hostname) but has its own internal filesystem (e.g. /.apptainer.d)
Tip: Isolate your host filesystem
To prevent accidental deletes/edits, add -c or -C flags to your apptainer commands to isolate filesystems:
$ apptainer shell -C ubuntu_latest.sif
Example: Pulling from NVIDIA GPU cloud (NGC)
NGC provides pre-built images for GPU accelerated applications. These images are large, so download them locally on your machine and then transfer to DAIC.
To install Apptainer locally, follow the official Installing Apptainer instructions.
Important: Cache and filesystem limits
By default, Apptainer images are saved to ~/.apptainer. To avoid quota issues, set the environment variable APPTAINER_CACHEDIR to a different location.
If you prefer (or need) a custom container image, you can build one from a definition file (*.def), that specifies your dependencies and setup steps.
On DAIC, you can build images directly if your current directory allows writes and sufficient quota (e.g., under staff-umbrella). For large or complex builds, it can be more convenient to build locally on your workstation and then transfer the resulting .sif file to DAIC.
Tip: Root privileges not always required
Apptainer supports rootless builds. You only need sudo when:
building from base images that require root setup (e.g., Bootstrap: docker on older systems), or
writing the resulting image to a protected location.
Otherwise, you can build directly:
$ apptainer build myimage.sif myimage.def
Example: CUDA-enabled container
An example definion file, cuda_based.def, for a cuda-enabled container may look as follows:
The header, specifies the source (eg, Bootstrap: docker) and the base image (From: nvidia/cuda:12.1.1-devel-ubuntu22.04). Here, the container builds on Ubuntu 22.04 with CUDA 12.1 pre-installed.
The rest of the file are optional data blobs or sections. In this example, the following blobs are used:
%post: the steps to download, configure and install needed custom software and libraries on the base image. In this example, the steps install git, clone a repo, and install a package via make
%runscript: the entry point to the container with the apptainer run command. In this example, the deviceQuery is executed once the container is run.
$cd /tudelft.net/staff-umbrella/<project>/apptainer
$ salloc --account=<your-account> --partition=all --cpus-per-task=2 --mem=1G --gres=gpu:1 --time=00:10:00
salloc: Granted job allocation 12345
$ srun apptainer run --nv -C cuda_based_image.sif
/cuda-samples/Samples/1_Utilities/deviceQuery/deviceQuery Starting...
CUDA Device Query (Runtime API) version (CUDART static linking)
Detected 1 CUDA Capable device(s)
Device 0: "NVIDIA L40"
CUDA Driver Version / Runtime Version 12.9 / 12.1
CUDA Capability Major/Minor version number: 8.9
Total amount of global memory: 46068 MBytes
...
deviceQuery, CUDA Driver = CUDART, CUDA Driver Version = 12.9, CUDA Runtime Version = 12.1, NumDevs = 1
Result = PASS
Tip: Enable GPU access
Always pass --nv to apptainer for GPU-accelerated workloads:
$ apptainer shell --nv -C cuda_based_image.sif
The host must have NVIDIA GPU drivers installed and the container must include CUDA dependencies.
Note on reproducibility
Definition-file builds are the most reproducible approach.
However, in cases of complex dependencies, you can first prototype interactively in writable sandbox mode first. In such cases, take note of all installation commands used in the sandbox, so you can include them in a recipe file. See Apptainer Sandbox Directories for more details.
Example: Extending existing images
During software development, it is common to incrementally build code and go through many iterations of debugging and testing.
To save time, you can base a new image on an existing one using the Bootstrap: localimage and From:<path/to/local/image> header.
This avoids re-installing the same dependencies with every iteration.
As an example, assume it is desirable to develop some code on the basis of the cuda_based.sif image created in the Example: CUDA-enabled container. Building from the original cuda_based.def file can take ~ 4 minutes. However, if the *.sif file is already available, building on top of it, via a dev_on_cuda_based.def file as below, takes ~ 2 minutes. This is already a time saving factor of 2.
As can be seen in this example, the new def file not only preserves the dependencies of the original image, but it also preserves a complete history of all build processes while giving flexible environment that can be customized as need arises.
Example: Deploying conda and pip in a container
There might be situations where you have a certain conda environment in your local machine that you need to set up in DAIC to commence your analysis. In such cases, deploying your conda environment in a container and sending this container to DAIC does the job for you.
As an example, let’s create a simple demo environment, environment.yml in our local machine,
Now, it is time to create the container definition file Apptainer.def. One option is to base the image on condaforge/miniforge, which is a minimal Ubuntu installation with conda preinstalled at /opt/conda:
Bootstrap: docker
From: condaforge/miniforge3:latest
%files
environment.yml /environment.yml
requirements.txt /requirements.txt
%post
# Update and install necessary packages apt-get update && apt-get install -y tree time vim ncdu speedtest-cli build-essential
# Create a new Conda environment using the environment files. mamba env create --quiet --file /environment.yml
# Clean up apt-get clean && rm -rf /var/lib/apt/lists/*
mamba clean --all -y
# Now add the script to activate the Conda environmentecho'. "/opt/conda/etc/profile.d/conda.sh"' >> $APPTAINER_ENVIRONMENTecho'conda activate apptainer' >> $APPTAINER_ENVIRONMENT
APPTAINER_ENVIRONMENT
The $APPTAINER_ENVIRONMENT variable in Apptainer refers to a special shell script that gets sourced when a container is run in shell mode. This is a key mechanism for setting up the environment for your container.
This adds a command to activate the “apptainer” Conda environment
This ensures your container automatically starts with the right environment activated
When a user runs your container with apptainer shell my-container.sif, these commands will execute automatically, ensuring:
The conda command is available
The “apptainer” environment is activated
All the Python packages specified in your environment.yml are available
This approach is much cleaner than requiring users to manually activate the environment every time they run the container. It makes your container more user-friendly and ensures consistent behavior.
This file is similar to the file in the Building images, with the addition of %files area. %files specifies the files in the host system (ie, your machine) that need to be copied to the container image, and optionally, where should they be available. In the previous example, the environment.yml file will be available in /opt/ in the container.
$ apptainer exec demo-env-image.sif which python
/opt/conda/envs/apptainer/bin/python
Perfect! This confirms that our container image built successfully and the Conda environment is automatically activated. The Python executable is correctly pointing to our custom environment path, indicating that all our dependencies should be available.
We are going to use the environment inside a container together with a Python script that we store outside the container.
Create the file analysis.py, which generate a plot:
$ apptainer exec demo-env-image.sif python analysis.py
$ ls
sine_wave.png
Warning
In the last example, the container read and wrote a file to the host system directly. This behavior is risky. You are strongly recommended to expose only the desired host directories to the container. See Exposing host directories
Exposing host directories
Depending on use case, it may be necessary for the container to read or write data from or to the host system. For example, to expose only files in a host directory called ProjectDataDir to the container image’s /mnt directory, add the --bind directive with appropriate <hostDir>:<containerDir> mapping to the commands you use to launch the container, in conjunction with the -C flag eg, shell or exec as below:
$ ls ProjectDataDir
raw_data.txt
$ apptainer shell -C --bind ProjectDataDir:/mnt ubuntu_latest.sif
Apptainer> ls /mnt
raw_data.txt
Apptainer> echo "Date: $(date)" >> /mnt/raw_data.txt
Apptainer> exit
$ tail -n1 ProjectDataDir/raw_data.txt
Date: Fri Mar 20 10:30:00 CET 2026
If the desire is to expose this directory as read-only inside the container, the --mount directive should be used instead of --bind, with rodesignation as follows:
Advanced: containers and (fake) native installation
It’s possible to use Apptainer to install and then use software as if it were installed natively in the host system. For example, if you are a bioinformatician, you may be using software like samtools or bcftools for many of your analyses, and it may be advantageous to call it directly. Let’s take this as an illustrative example:
Create a directory structure: an exec directory for container images and a bin directory for symlinks:
Learn the Vim text editor for efficient file editing on DAIC.
What you’ll learn
By the end of this tutorial, you’ll be able to:
Open, edit, save, and quit files in Vim
Navigate efficiently without touching the mouse
Delete, copy, and paste text
Search and replace
Edit SLURM scripts and Python code on the cluster
Time: About 30 minutes
Prerequisites: Basic familiarity with command line. Complete Bash Basics first if you’re new to Linux.
Why learn Vim?
When working on DAIC, you’ll often need to edit files directly on the cluster - tweaking a batch script, fixing a bug in your code, or checking a configuration file. Since DAIC is accessed via SSH (no graphical interface), you need a terminal-based text editor.
Vim is the most powerful and ubiquitous terminal editor. It’s installed on every Linux system, so the skills you learn transfer everywhere. While Vim has a steeper learning curve than simpler editors like nano, investing time to learn it pays off:
Speed: Once fluent, you can edit text faster than with any other editor
Availability: Always there, no installation needed
Efficiency: Designed to minimize hand movement and keystrokes
Ubiquity: Same editor on your laptop, on DAIC, on any server
This tutorial teaches you enough Vim to be comfortable editing files on DAIC. You don’t need to master everything - even basic Vim skills will serve you well.
The most important thing: how to quit
Before anything else, let’s address the most common Vim problem: getting stuck. If you accidentally open Vim and don’t know how to exit, here’s what to do:
Press Esc several times (ensures you’re in the right mode)
Type :q! and press Enter
This quits without saving. If you want to save your changes first, use :wq instead.
Command
What it does
:q
Quit (only works if no unsaved changes)
:q!
Quit and discard changes
:w
Save the file
:wq
Save and quit
ZZ
Shortcut for save and quit
Now that you know how to escape, let’s learn how to actually use Vim.
Understanding Vim’s philosophy
Vim works differently from editors you may be used to (like Word, VS Code, or even Notepad). The key insight is:
You spend more time navigating and editing text than typing new text.
Think about it: when you edit code, most of your time is spent reading, moving around, deleting lines, copying blocks, and making small changes. Typing fresh text is a small fraction of editing.
Vim is optimized for this reality. Instead of always being ready to type (like most editors), Vim has different modes for different tasks:
Normal mode: Navigate and manipulate text (where you spend most time)
Insert mode: Type new text
Visual mode: Select text
Command mode: Run commands
This might feel awkward at first, but it’s what makes Vim so efficient.
Modes explained
Normal mode: your home base
When you open Vim, you’re in Normal mode. This is your home base - you’ll return here constantly.
In Normal mode, every key is a command:
j moves down (not typing the letter “j”)
dd deletes a line
w jumps to the next word
You cannot type text in Normal mode. This is intentional - it lets every key be a powerful command instead of just inserting a character.
To return to Normal mode from anywhere, press Esc. If you’re ever confused about what mode you’re in, press Esc a few times. You’ll always end up in Normal mode.
Insert mode: typing text
When you need to type new text, you enter Insert mode. The most common way is pressing i (for “insert”).
In Insert mode:
You can type normally, like any other editor
The bottom of the screen shows -- INSERT --
Backspace, arrow keys, and Enter work as expected
When done typing, press Esc to return to Normal mode.
There are several ways to enter Insert mode, each starting you in a different position:
Key
Where you start typing
i
Before the cursor
a
After the cursor
I
At the beginning of the line
A
At the end of the line
o
On a new line below
O
On a new line above
The most common are i (insert here), A (append to line), and o (open new line).
Visual mode: selecting text
Visual mode lets you select text, similar to clicking and dragging in other editors. Press v to enter Visual mode, then move the cursor to extend the selection.
Once you’ve selected text, you can:
Press d to delete it
Press y to copy (“yank”) it
Press > to indent it
Press Esc to cancel the selection and return to Normal mode.
Command mode: running commands
Press : to enter Command mode. You’ll see a colon appear at the bottom of the screen, where you can type commands like:
:w - save (write) the file
:q - quit
:set number - show line numbers
:%s/old/new/g - find and replace
Press Enter to execute the command, or Esc to cancel.
Your first Vim session
Let’s put this together with a hands-on exercise. We’ll create a simple Python script.
Step 1: Open Vim
$ vim hello.py
You’re now in Vim, looking at an empty file. Notice:
The cursor is at the top left
Tildes (~) mark empty lines beyond the file
The bottom shows the filename
You’re in Normal mode. If you try typing, nothing will appear (or unexpected things will happen).
Step 2: Enter Insert mode and type
Press i. The bottom of the screen now shows -- INSERT --.
Type this code:
#!/usr/bin/env python3print("Hello from DAIC!")
Step 3: Return to Normal mode
Press Esc. The -- INSERT -- message disappears. You’re back in Normal mode.
Step 4: Save and quit
Type :wq and press Enter.
You’ve saved the file and exited Vim. Verify it worked:
$ cat hello.py
#!/usr/bin/env python3
print("Hello from DAIC!")
$ python hello.py
Hello from DAIC!
Congratulations - you’ve completed your first Vim edit!
Navigation: moving around efficiently
One of Vim’s superpowers is fast navigation. In Normal mode, you can move around without touching the mouse or arrow keys.
Basic movement: hjkl
The home row keys h, j, k, l move the cursor:
k
↑
h ← → l
↓
j
h - left
j - down (think: “j” hangs down below the line)
k - up
l - right
Arrow keys also work, but hjkl keeps your hands on the home row. It feels strange at first but becomes natural with practice.
Moving by words
Character-by-character movement is slow. Jump by words instead:
Key
Movement
w
Forward to start of next word
b
Backward to start of previous word
e
Forward to end of current/next word
Try it: open a file and press w repeatedly. Watch the cursor hop from word to word.
Moving within a line
Key
Movement
0
Beginning of line (column zero)
^
First non-blank character
$
End of line
The ^ and $ symbols come from regular expressions, where they mean start and end.
Moving through the file
Key
Movement
gg
First line of file
G
Last line of file
42G
Line 42 (any number works)
Ctrl+d
Down half a page
Ctrl+u
Up half a page
Ctrl+f
Forward one page
Ctrl+b
Backward one page
When reviewing a log file, G takes you straight to the end (most recent output), and gg takes you back to the beginning.
Practice exercise
Open any file:
$ vim /etc/passwd
Now practice:
Press G to go to the last line
Press gg to go to the first line
Press 10G to go to line 10
Press $ to go to the end of the line
Press 0 to go to the beginning
Press w several times to move by words
Press :q to quit (no need to save - you shouldn’t modify this file)
Editing text
Now that you can navigate, let’s learn to edit.
Deleting text
In Normal mode, d is the delete command. It combines with movement:
Command
What it deletes
x
Character under cursor
dd
Entire line
dw
From cursor to start of next word
de
From cursor to end of word
d$
From cursor to end of line
d0
From cursor to beginning of line
dG
From current line to end of file
dgg
From current line to beginning of file
The pattern is: d + movement. The dd (delete line) is used so often it gets a shortcut.
Undo and redo
Made a mistake? No problem:
Command
Action
u
Undo last change
Ctrl+r
Redo (undo the undo)
Vim remembers many levels of undo, so you can press u repeatedly to go back through history.
Copying and pasting
In Vim, copying is called “yanking” (the y key). Pasting is “putting” (the p key).
Command
Action
yy
Yank (copy) the current line
yw
Yank from cursor to start of next word
y$
Yank from cursor to end of line
p
Put (paste) after cursor
P
Put before cursor
The pattern is similar to delete: y + movement.
Here’s a useful trick: when you delete with d, the deleted text is saved (like “cut” in other editors). So dd followed by p moves a line - delete it, then paste it elsewhere.
Changing text
The c command deletes and puts you in Insert mode - useful for replacing text:
Command
Action
cw
Change word (delete word, enter Insert mode)
cc
Change entire line
c$
Change to end of line
This is faster than deleting and then inserting separately.
Repeating actions
One of Vim’s best features: press . to repeat the last change.
Example workflow:
Find a line you want to delete: /TODO
Delete it: dd
Find the next one: n
Repeat the deletion: .
Continue: n, ., n, ., …
Searching
Finding text
To search forward, press /, type your search term, and press Enter:
/error
Vim jumps to the first match. Then:
n - next match
N - previous match
To search backward, use ? instead of /.
To search for the word under your cursor, press * (forward) or # (backward).
Find and replace
To replace text, use the substitute command:
:s/old/new/
This replaces the first occurrence of “old” with “new” on the current line.
Add flags for more control:
Command
What it does
:s/old/new/g
Replace all occurrences on current line
:%s/old/new/g
Replace all occurrences in entire file
:%s/old/new/gc
Replace all, but ask for confirmation each time
The % means “entire file” and g means “global” (all occurrences, not just the first).
Example - update a variable name throughout your code:
:%s/learning_rate/lr/g
Visual mode: selecting text
Sometimes you need to select a region of text before acting on it. Visual mode lets you see exactly what you’re selecting before you delete, copy, or modify it.
Three types of selection
Vim offers three selection styles for different situations:
Character selection (v) - Select specific characters, like highlighting with a mouse. Use when you need part of a line.
Line selection (V) - Select entire lines at once. Use when working with whole lines of code - which is most of the time.
Block selection (Ctrl+v) - Select a rectangular region. Use for columnar data or adding text to multiple lines.
Line selection (V) - the most useful
Line selection is what you’ll use most often when editing code. It selects complete lines, which is usually what you want.
Example: Delete a function
You have a Python file and want to delete an entire function:
You have space-separated data and want to remove the second column:
apple red 5
banana yellow 3
grape purple 8
Steps:
Move to the r in red
Press Ctrl+v
Press 2j to extend down
Press e to extend to end of word
Press d to delete
Result:
apple 5
banana 3
grape 8
Quick reference
Key
When to use
V
Deleting, copying, or indenting whole lines (most common)
v
Selecting part of a line
Ctrl+v
Editing columns or multiple lines at once
After selecting, these actions work on your selection:
d - delete
y - yank (copy)
c - change (delete and start typing)
> - indent
< - unindent
: - run a command on selected lines
Practical workflows for DAIC
Editing a batch script
You need to change the time limit in your SLURM script:
$ vim submit.sh
Search for the time directive: /time
Press n until you find #SBATCH --time=1:00:00
Move to the “1”: f1 (find the character “1”)
Change the number: cw then type 4 then Esc
Save and quit: :wq
Adding a line to a script
You need to add a new SBATCH directive:
$ vim submit.sh
Navigate to the SBATCH section: /SBATCH
Open a new line below: o
Type: #SBATCH --gres=gpu:1
Exit insert mode: Esc
Save and quit: :wq
Viewing a log file
Check the output of a completed job:
$ vim slurm_12345.out
Go to the end (most recent output): G
Search backward for errors: ?error
Quit without saving: :q
For just viewing, you could also use less slurm_12345.out, but Vim’s search is more powerful.
Copying code between files
You need to copy a function from one file to another:
$ vim model.py
Find the function: /def train
Start Visual line selection: V
Select the entire function (move down): } (jumps to next blank line)
Yank (copy): y
Open the other file: :e utils.py
Navigate to where you want the function
Paste: p
Save: :w
Go back: :e model.py or Ctrl+^
Configuring Vim
Vim reads settings from ~/.vimrc when it starts. Here’s a good starting configuration:
$ vim ~/.vimrc
Enter Insert mode (i) and add:
" Line numberssetnumber" Syntax highlightingsyntaxon" Indentationsettabstop=4" Tab widthsetshiftwidth=4" Indent widthsetexpandtab" Use spaces, not tabssetautoindent" Copy indent from previous line" Searchsetignorecase" Case-insensitive searchsetsmartcase" ...unless you use capitalssethlsearch" Highlight matchessetincsearch" Search as you type" Usabilitysetshowmatch" Highlight matching bracketssetmouse=a" Enable mousesetruler" Show cursor positionsetwildmenu" Better command completion" Colorssetbackground=darkcolorschemedesert
Lines starting with " are comments. Save with :wq and the settings apply next time you open Vim.
Learning more
This tutorial covers the essentials. To go further:
Built-in tutorial: Run vimtutor in your terminal for an interactive 30-minute lesson:
$ vimtutor
Gradual learning: Don’t try to learn everything at once. Start with:
i to insert, Esc to stop
:wq to save and quit
dd to delete lines, u to undo
Then gradually add new commands as the basic ones become automatic.
Practice: The only way to get comfortable with Vim is to use it. Force yourself to use it for small edits, and the commands will become muscle memory.
Cheat sheet
Modes
Key
Mode
Esc
Normal (command) mode
i, a, o
Insert mode
v, V
Visual mode
:
Command mode
Essential commands
Command
Action
:w
Save
:q
Quit
:wq
Save and quit
:q!
Quit without saving
u
Undo
Ctrl+r
Redo
Movement
Key
Movement
h j k l
Left, down, up, right
w, b
Forward, backward by word
0, $
Beginning, end of line
gg, G
Beginning, end of file
/pattern
Search forward
Editing
Command
Action
i
Insert before cursor
a
Insert after cursor
o
Insert on new line below
dd
Delete line
yy
Copy line
p
Paste
cw
Change word
.
Repeat last change
Summary
You’ve learned the essential Vim workflow:
Task
Commands
Open a file
vim filename
Enter insert mode
i, a, o
Return to normal mode
Esc
Save
:w
Quit
:q or :wq
Navigate
hjkl, w, b, gg, G
Delete
x, dd, dw
Copy/paste
yy, p
Undo/redo
u, Ctrl+r
Search
/pattern, n, N
Replace
:%s/old/new/g
Select lines
V + movement
Exercises
Practice these tasks to build muscle memory:
Exercise 1: Basic editing
Create a new file, add three lines of text, save and quit. Then reopen it and verify your changes.
Check your work
After :wq, verify with:
$ cat myfile.txt
line one
line two
line three
If the file is empty, you may have quit without saving (:q! instead of :wq).
Exercise 2: Navigation
Open a Python file and practice: go to end (G), go to beginning (gg), jump by words (w, b), go to specific line (10G).
Check your work
Check your position with :set number to show line numbers. After G, you should be on the last line. After gg, you should be on line 1. After 10G, you should be on line 10.
Exercise 3: Delete and undo
Open a file, delete a line (dd), undo (u), delete a word (dw), undo again.
Check your work
After each u, the deleted content should reappear. If undo doesn’t work, make sure you’re in Normal mode (press Esc first).
Exercise 4: Copy and paste
Copy a line (yy), move to a new location, paste it (p). Then try with multiple lines using V.
Check your work
After yy and p, you should see the same line duplicated. With V, select multiple lines (they highlight), then y to copy and p to paste them elsewhere.
Exercise 5: Search and replace
Open a file and search for a word (/word). Then replace all occurrences of one word with another (:%s/old/new/g).
Check your work
After /word and pressing Enter, the cursor jumps to the first match. Press n to see subsequent matches. After :%s/old/new/g, Vim reports how many substitutions were made (e.g., “5 substitutions on 3 lines”).
Exercise 6: Real task
Edit a SLURM batch script: change the time limit, add a new #SBATCH directive, and save.
DAIC GPU nodes have GPUs on different NUMA nodes (CPU sockets). You must set NCCL_P2P_DISABLE=1 in your job scripts for multi-GPU training to work. See NCCL Configuration below.
What you’ll learn
By the end of this tutorial, you’ll be able to:
Understand when and why to use multiple GPUs
Train models across GPUs with PyTorch Lightning
Use native PyTorch Distributed Data Parallel (DDP)
Scale training with Hugging Face Accelerate
Configure Slurm jobs for multi-GPU and multi-node training
This tutorial includes complete, runnable example scripts in the examples/ directory. Copy them to your project storage and test on DAIC:
examples/lightning/ - PyTorch Lightning example
examples/ddp/ - Native PyTorch DDP example
examples/accelerate/ - Hugging Face Accelerate example
When to use multiple GPUs
Training on multiple GPUs makes sense when:
Training is slow: A single GPU takes hours or days per epoch
Model fits in memory: The model fits on one GPU, but you want faster training
Large batch sizes: You need larger effective batch sizes for better convergence
Multiple GPUs do not help when:
Your model doesn’t fit on a single GPU (you need model parallelism instead)
Data loading is the bottleneck
Training is already fast (communication overhead may slow things down)
The dataset is small (like MNIST) - GPU communication overhead exceeds computation time
About the examples
The examples use CIFAR-10 with ResNet18, which is large enough to demonstrate multi-GPU speedup (~1.3x with 2 GPUs). For production workloads with larger models and datasets, expect near-linear scaling.
Scaling strategies
Strategy
What it does
When to use
Data Parallel
Same model on each GPU, different data batches
Most common, covered here
Model Parallel
Model split across GPUs
Very large models (LLMs)
Pipeline Parallel
Model layers on different GPUs
Very deep networks
This tutorial focuses on data parallelism - the most common and easiest approach.
How data parallelism works
The model is replicated on each GPU
Each GPU processes a different batch of data
Gradients are synchronized across GPUs
Weights are updated identically on all GPUs
With 2 GPUs and batch size 32 per GPU, you effectively train with batch size 64.
Part 1: PyTorch Lightning
PyTorch Lightning is the easiest way to scale training. It handles distributed training automatically - you write single-GPU code, Lightning handles the rest.
# src/train.pyimporttorchimporttorch.nnasnnimporttorch.nn.functionalasFimportlightningasLfromtorch.utils.dataimportDataLoader,random_splitfromtorchvisionimportdatasets,transformsclassImageClassifier(L.LightningModule):def__init__(self,learning_rate=1e-3):super().__init__()self.save_hyperparameters()self.model=nn.Sequential(nn.Flatten(),nn.Linear(28*28,256),nn.ReLU(),nn.Dropout(0.2),nn.Linear(256,128),nn.ReLU(),nn.Dropout(0.2),nn.Linear(128,10))defforward(self,x):returnself.model(x)deftraining_step(self,batch,batch_idx):x,y=batchlogits=self(x)loss=F.cross_entropy(logits,y)acc=(logits.argmax(dim=1)==y).float().mean()self.log('train_loss',loss,prog_bar=True)self.log('train_acc',acc,prog_bar=True)returnlossdefvalidation_step(self,batch,batch_idx):x,y=batchlogits=self(x)loss=F.cross_entropy(logits,y)acc=(logits.argmax(dim=1)==y).float().mean()self.log('val_loss',loss,prog_bar=True)self.log('val_acc',acc,prog_bar=True)defconfigure_optimizers(self):returntorch.optim.Adam(self.parameters(),lr=self.hparams.learning_rate)classMNISTDataModule(L.LightningDataModule):def__init__(self,data_dir='./data',batch_size=64,num_workers=4):super().__init__()self.data_dir=data_dirself.batch_size=batch_sizeself.num_workers=num_workersself.transform=transforms.Compose([transforms.ToTensor(),transforms.Normalize((0.1307,),(0.3081,))])defprepare_data(self):# Download (runs on rank 0 only)datasets.MNIST(self.data_dir,train=True,download=True)datasets.MNIST(self.data_dir,train=False,download=True)defsetup(self,stage=None):ifstage=='fit'orstageisNone:mnist_full=datasets.MNIST(self.data_dir,train=True,transform=self.transform)self.mnist_train,self.mnist_val=random_split(mnist_full,[55000,5000])ifstage=='test'orstageisNone:self.mnist_test=datasets.MNIST(self.data_dir,train=False,transform=self.transform)deftrain_dataloader(self):returnDataLoader(self.mnist_train,batch_size=self.batch_size,shuffle=True,num_workers=self.num_workers,persistent_workers=True)defval_dataloader(self):returnDataLoader(self.mnist_val,batch_size=self.batch_size,num_workers=self.num_workers,persistent_workers=True)defmain():# Datadatamodule=MNISTDataModule(data_dir='/tudelft.net/staff-umbrella/<project>/data',batch_size=64,num_workers=4)# Modelmodel=ImageClassifier(learning_rate=1e-3)# Trainer - single GPUtrainer=L.Trainer(max_epochs=10,accelerator='gpu',devices=1,precision='16-mixed',enable_progress_bar=True,)trainer.fit(model,datamodule)if__name__=='__main__':main()
Scaling to multiple GPUs
The only change needed is in the Trainer configuration:
# Multi-GPU: use all available GPUs on one nodetrainer=L.Trainer(max_epochs=10,accelerator='gpu',devices=2,# Use 2 GPUsstrategy='ddp',# Distributed Data Parallelprecision='16-mixed',)
That’s it. Lightning handles:
Spawning processes for each GPU
Distributing data across GPUs
Synchronizing gradients
Logging from rank 0 only
Slurm job script for multi-GPU
#!/bin/bash
#SBATCH --account=<your-account>#SBATCH --partition=all#SBATCH --time=2:00:00#SBATCH --nodes=1#SBATCH --ntasks-per-node=1#SBATCH --cpus-per-task=8#SBATCH --mem=64G#SBATCH --gres=gpu:2#SBATCH --output=train_%j.outmodule purge
module load 2025/gpu cuda/12.9
cd /tudelft.net/staff-umbrella/<project>/lightning-multi-gpu
# Set number of workers based on CPUsexportNUM_WORKERS=$((SLURM_CPUS_PER_TASK /4))srun uv run python src/train.py
Key points:
--gres=gpu:2: Request 2 GPUs
--cpus-per-task=8: Enough CPUs for data loading (4 per GPU)
--ntasks-per-node=1: Lightning spawns its own processes
Multi-node training
Scale beyond one machine with minimal changes:
trainer=L.Trainer(max_epochs=10,accelerator='gpu',devices=2,# GPUs per nodenum_nodes=2,# Number of nodesstrategy='ddp',precision='16-mixed',)
Slurm script for multi-node:
#!/bin/bash
#SBATCH --account=<your-account>#SBATCH --partition=all#SBATCH --time=4:00:00#SBATCH --nodes=2#SBATCH --ntasks-per-node=1#SBATCH --cpus-per-task=8#SBATCH --mem=64G#SBATCH --gres=gpu:2#SBATCH --output=train_%j.outmodule purge
module load 2025/gpu cuda/12.9
cd /tudelft.net/staff-umbrella/<project>/lightning-multi-gpu
# Get master address from first nodeexportMASTER_ADDR=$(scontrol show hostnames $SLURM_JOB_NODELIST| head -n 1)exportMASTER_PORT=29500srun uv run python src/train.py
Exercise 1: Scale with Lightning
Create the Lightning project above
Train on 1 GPU and note the time per epoch
Change to 2 GPUs and compare
Verify both runs achieve similar accuracy
Check your work
Both configurations should achieve ~97% validation accuracy. Note that for MNIST, you may not see a speedup - the dataset is too small and communication overhead dominates. With larger datasets and models, you would see near-linear scaling.
Part 2: PyTorch DDP (native)
If you need more control or can’t use Lightning, PyTorch’s DistributedDataParallel (DDP) is the native approach.
Key concepts
World size: Total number of processes (GPUs)
Rank: Unique ID for each process (0 to world_size-1)
Local rank: GPU index on the current node (0 to GPUs_per_node-1)
DDP training script
# src/train_ddp.pyimportosimporttorchimporttorch.nnasnnimporttorch.nn.functionalasFimporttorch.distributedasdistfromtorch.nn.parallelimportDistributedDataParallelasDDPfromtorch.utils.dataimportDataLoaderfromtorch.utils.data.distributedimportDistributedSamplerfromtorchvisionimportdatasets,transformsdefsetup():"""Initialize distributed training."""dist.init_process_group(backend='nccl')torch.cuda.set_device(int(os.environ['LOCAL_RANK']))defcleanup():"""Clean up distributed training."""dist.destroy_process_group()defget_rank():returndist.get_rank()defis_main_process():returnget_rank()==0classSimpleNet(nn.Module):def__init__(self):super().__init__()self.flatten=nn.Flatten()self.fc1=nn.Linear(28*28,256)self.fc2=nn.Linear(256,128)self.fc3=nn.Linear(128,10)self.dropout=nn.Dropout(0.2)defforward(self,x):x=self.flatten(x)x=F.relu(self.fc1(x))x=self.dropout(x)x=F.relu(self.fc2(x))x=self.dropout(x)returnself.fc3(x)deftrain_epoch(model,loader,optimizer,device):model.train()total_loss=0correct=0total=0forbatch_idx,(data,target)inenumerate(loader):data,target=data.to(device),target.to(device)optimizer.zero_grad()output=model(data)loss=F.cross_entropy(output,target)loss.backward()optimizer.step()total_loss+=loss.item()pred=output.argmax(dim=1)correct+=pred.eq(target).sum().item()total+=target.size(0)returntotal_loss/len(loader),correct/totaldefvalidate(model,loader,device):model.eval()total_loss=0correct=0total=0withtorch.no_grad():fordata,targetinloader:data,target=data.to(device),target.to(device)output=model(data)total_loss+=F.cross_entropy(output,target).item()pred=output.argmax(dim=1)correct+=pred.eq(target).sum().item()total+=target.size(0)returntotal_loss/len(loader),correct/totaldefmain():# Initialize distributedsetup()local_rank=int(os.environ['LOCAL_RANK'])device=torch.device(f'cuda:{local_rank}')# Datatransform=transforms.Compose([transforms.ToTensor(),transforms.Normalize((0.1307,),(0.3081,))])train_dataset=datasets.MNIST('/tudelft.net/staff-umbrella/<project>/data',train=True,download=False,transform=transform)val_dataset=datasets.MNIST('/tudelft.net/staff-umbrella/<project>/data',train=False,download=False,transform=transform)# Distributed sampler ensures each GPU gets different datatrain_sampler=DistributedSampler(train_dataset,shuffle=True)val_sampler=DistributedSampler(val_dataset,shuffle=False)train_loader=DataLoader(train_dataset,batch_size=64,sampler=train_sampler,num_workers=4,pin_memory=True)val_loader=DataLoader(val_dataset,batch_size=64,sampler=val_sampler,num_workers=4,pin_memory=True)# Model - wrap in DDPmodel=SimpleNet().to(device)model=DDP(model,device_ids=[local_rank])optimizer=torch.optim.Adam(model.parameters(),lr=1e-3)# Training loopforepochinrange(10):# Important: set epoch for proper shufflingtrain_sampler.set_epoch(epoch)train_loss,train_acc=train_epoch(model,train_loader,optimizer,device)val_loss,val_acc=validate(model,val_loader,device)# Only print from main processifis_main_process():print(f'Epoch {epoch+1}: 'f'train_loss={train_loss:.4f}, train_acc={train_acc:.4f}, 'f'val_loss={val_loss:.4f}, val_acc={val_acc:.4f}')# Save model (only from main process)ifis_main_process():torch.save(model.module.state_dict(),'model.pt')print('Model saved to model.pt')cleanup()if__name__=='__main__':main()
Key differences from single-GPU
Initialize process group: dist.init_process_group()
Wrap model in DDP: model = DDP(model, device_ids=[local_rank])
Use DistributedSampler: Ensures each GPU gets different data
Set sampler epoch: train_sampler.set_epoch(epoch) for proper shuffling
Save from rank 0 only: Avoid file conflicts
Access original model: Use model.module when saving
Note: --ntasks-per-node=4 launches 4 processes, one per GPU.
Exercise 2: Native DDP
Create the DDP training script
Run with 2 GPUs using torchrun
Verify the DistributedSampler splits data correctly
Check your work
Each GPU should process half the data:
# With 60000 training samples and 2 GPUs:
# Each GPU sees 30000 samples per epoch
GPU 0: Processing batches 0-468
GPU 1: Processing batches 0-468
Part 3: Hugging Face Accelerate
Accelerate provides a middle ground - more control than Lightning, less boilerplate than raw DDP.
Setup
$ uv add accelerate transformers datasets
Accelerate training script
# src/train_accelerate.pyimporttorchimporttorch.nnasnnimporttorch.nn.functionalasFfromtorch.utils.dataimportDataLoaderfromtorchvisionimportdatasets,transformsfromaccelerateimportAcceleratorclassSimpleNet(nn.Module):def__init__(self):super().__init__()self.flatten=nn.Flatten()self.fc1=nn.Linear(28*28,256)self.fc2=nn.Linear(256,128)self.fc3=nn.Linear(128,10)defforward(self,x):x=self.flatten(x)x=F.relu(self.fc1(x))x=F.relu(self.fc2(x))returnself.fc3(x)defmain():# Initialize acceleratoraccelerator=Accelerator(mixed_precision='fp16')# Datatransform=transforms.Compose([transforms.ToTensor(),transforms.Normalize((0.1307,),(0.3081,))])train_dataset=datasets.MNIST('/tudelft.net/staff-umbrella/<project>/data',train=True,download=False,transform=transform)train_loader=DataLoader(train_dataset,batch_size=64,shuffle=True,num_workers=4)# Model and optimizermodel=SimpleNet()optimizer=torch.optim.Adam(model.parameters(),lr=1e-3)# Prepare for distributed trainingmodel,optimizer,train_loader=accelerator.prepare(model,optimizer,train_loader)# Training loopforepochinrange(10):model.train()total_loss=0forbatch_idx,(data,target)inenumerate(train_loader):optimizer.zero_grad()output=model(data)loss=F.cross_entropy(output,target)accelerator.backward(loss)optimizer.step()total_loss+=loss.item()# Print from main process onlyifaccelerator.is_main_process:print(f'Epoch {epoch+1}: loss={total_loss/len(train_loader):.4f}')# Save modelaccelerator.wait_for_everyone()ifaccelerator.is_main_process:unwrapped_model=accelerator.unwrap_model(model)torch.save(unwrapped_model.state_dict(),'model.pt')if__name__=='__main__':main()
Key features
Minimal code changes: Just wrap with accelerator.prepare()
Automatic device placement: No manual .to(device)
Mixed precision: Built-in with mixed_precision='fp16'
Gradient accumulation: Easy with accumulate() context
Data loading often becomes the bottleneck with multiple GPUs.
Tips:
Use num_workers proportional to CPUs: typically 4 workers per GPU
Enable pin_memory=True for faster GPU transfer
Use persistent_workers=True to avoid worker restart overhead
Store data on fast storage (SSD/NVMe when available)
DataLoader(dataset,batch_size=64,num_workers=4,# Per GPUpin_memory=True,# Faster transfer to GPUpersistent_workers=True,# Keep workers aliveprefetch_factor=2,# Batches to prefetch per worker)
Batch size scaling
When using N GPUs, you have options:
Keep per-GPU batch size: Effective batch = N * per_GPU_batch
Faster training, may need learning rate adjustment
Keep total batch size: per_GPU_batch = total / N
Same training dynamics, just faster
Learning rate scaling rule: When increasing batch size by factor K, increase learning rate by factor K (or sqrt(K) for more conservative scaling).
# Example: scaling from 1 to 2 GPUsbase_lr=1e-3base_batch=64num_gpus=2# Linear scalingscaled_lr=base_lr*num_gpus# 4e-3
DAIC GPU nodes have GPUs distributed across multiple NUMA nodes (CPU sockets). The GPUs communicate via the QPI/UPI interconnect rather than NVLink, which requires specific NCCL configuration.
Required settings
Add these environment variables to your job scripts:
# Required: Disable P2P (peer-to-peer) communication# P2P doesn't work between GPUs on different NUMA nodesexportNCCL_P2P_DISABLE=1
Why this is needed
Check GPU topology on a compute node:
$ nvidia-smi topo -m
GPU0 GPU1 CPU Affinity NUMA Affinity
GPU0 X SYS 16-17 2
GPU1 SYS X 32-33 4
The SYS connection means GPUs communicate through the CPU interconnect (QPI/UPI), not direct P2P. Without NCCL_P2P_DISABLE=1, NCCL attempts P2P transfers that hang.
Performance expectations
With NCCL_P2P_DISABLE=1 on DAIC:
Configuration
ResNet18 on CIFAR-10
Speedup
1 GPU
7.8s/epoch
baseline
2 GPUs
6.1s/epoch
1.28x
The speedup is less than 2x because communication goes through CPU memory. Larger models and datasets see better scaling.
Part 5: Troubleshooting
Training hangs with multiple GPUs
Training hangs after “Initializing distributed” or “All distributed processes registered”.
Cause: NCCL P2P communication fails between GPUs on different NUMA nodes.
Solution:
exportNCCL_P2P_DISABLE=1
NCCL errors
NCCL error: unhandled system error
Causes:
Network issues between nodes
Mismatched CUDA/NCCL versions
Firewall blocking ports
Solutions:
# Use shared memory for single-nodeexportNCCL_SHM_DISABLE=0# Debug loggingexportNCCL_DEBUG=INFO
# Specify network interfaceexportNCCL_SOCKET_IFNAME=eth0
#SBATCH --gres=gpu:2 # Number of GPUs#SBATCH --cpus-per-task=8 # CPUs for data loading#SBATCH --ntasks-per-node=1 # For Lightning#SBATCH --ntasks-per-node=4 # For DDP/torchrun